The Codon Stamp
Three substrate observables from one set of per-base inputs

A codon is three stacked aromatic bases — six toroidal vortices for purines and pyrimidines on a 6.8 Å column twisted 34.3° per step. The channel-with-memory and nested-modon sections set up the substrate framework’s reading of this column as a coherent 3D stamp on the substrate’s flow geometry, not just three written letters. If that picture is right, the 64 codons are 64 distinct stamps with a measurable similarity relation among them, and synonymous codons (codons that translate to the same amino acid) carry different stamps despite sharing an amino-acid label.
This section builds the stamp explicitly. The per-base aromatic-vortex profiles from Aromatic Rings as Toroidal Vortices define the alphabet; convolving three of them on the polar axis gives the codon’s 3D substrate fingerprint; an L2 metric on that space gives the 64×64 distance matrix. The same per-base inputs feed two other observables — pair-level Watson-Crick binding (built up in the base-pairing section) and the 64×64 codon-anticodon cognate-recognition matrix. One set of inputs; one overall scaling factor; three independent classes of substrate prediction.
The per-base vortex profile
Aromatic-rings physics treats each π-aromatic ring as a closed-loop substrate raceway above and below the molecular plane. For each of the five nucleic-acid bases (A, G, C, T, U), this gives one (pyrimidines) or two (purines, with fused 6-5 rings) toroidal vortices with a definite spatial profile of substrate displacement:
\phi_B(\vec{r}) \;\equiv\; \text{substrate-displacement field of base } B,\;\text{centered at ring centroid, in the molecular frame.}
What goes into \phi_B in practice:
| Input | Source | Status |
|---|---|---|
| Ring-current strength | NICS(0), NICS(1), NICS_zz scans (DFT) | Published for all five bases; methods vary, ~0.5 ppm scatter |
| π-electron density profile | Computed electron density (B3LYP or higher) | Published |
| Heteroatom positions | Crystal structure | Standard |
| Static dipole moment | Spectroscopy or DFT | Published |
| Aromatic-vortex coupling strength | Framework parameter | One overall scaling factor |
The pyrimidines (C, T, U) each carry one toroidal vortex over their 6-electron ring. The purines (A, G) carry a more complex profile: a 10-electron ring current distributed over the fused 6-5 system, with the dominant contribution from the larger ring and a secondary lobe over the 5-membered ring. Standard NICS work (originated by Schleyer and refined by many groups since) places purines as more aromatic than pyrimidines, with within-class orderings that depend on basis set and probe location — orderings that require primary-source verification before they enter a quantitative calculation.
Stacking three bases into a codon stamp
A codon is three stacked bases. The stacking geometry depends on context:
- In B-DNA storage: rise h \approx 3.4 Å, twist \Delta\theta \approx 34.3°/\text{bp} (the 10.5 bp/turn pitch developed in B-DNA’s Pitch from the Packing Fraction).
- At the ribosomal A-site: the codon-anticodon mini-duplex is in A-form RNA geometry, with h \approx 2.8 Å and \Delta\theta \approx 32.7°/\text{bp}.
Both geometries appear in the cellular lifecycle of any codon. The calculations below use B-DNA values for the codon-distance matrix (the storage form where the stamp persists between expression events) and A-form values for the codon-anticodon binding matrix (the reading form at the ribosome).
The codon stamp is
\Phi_C(\vec{r}) \;=\; \sum_{n=0}^{2}\,R_{n\Delta\theta}\;\phi_{B_n}\!\bigl(\vec{r}-n\,h\,\hat{z}\bigr)
where C = B_0 B_1 B_2 is the codon read 5' \to 3', \hat{z} is the polar axis, and R_{n\Delta\theta} is rotation about \hat{z}. Off-axis, \Phi_C(\vec{r}) is a 3D function with three peaks at z = 0,\,h,\,2h, each rotated relative to the last, each carrying the in-plane fingerprint of its base. A codon’s stamp is therefore a function on \mathbb{R}^3, not a number — and the substrate’s claim is that this function (not the amino-acid label) is what the next residue’s local field sees as the peptide bond forms.
Distance on stamp space
The natural metric is L2 on the displacement-field space, normalized so identical codons sit at distance 0:
d(C_1, C_2) \;=\; \left[\frac{\int|\Phi_{C_1}-\Phi_{C_2}|^2\,d^3r}{\tfrac{1}{2}\!\int(|\Phi_{C_1}|^2+|\Phi_{C_2}|^2)\,d^3r}\right]^{1/2}.
It is symmetric, scale-invariant, and identically zero on coincident stamps. The equivalent cosine form
d_\text{cos}(C_1, C_2) \;=\; 1 - \frac{\langle \Phi_{C_1},\,\Phi_{C_2}\rangle}{\|\Phi_{C_1}\|\,\|\Phi_{C_2}\|}
reads as a similarity score and is easier to relate to existing codon-usage correlation tooling.
Two simplifications fall out of the convolution structure:
Single-position substitutions (e.g., CUU vs CUC, differing only at position 3): the integral over the unchanged positions cancels, and the distance reduces to a per-position contribution determined by the difference of two per-base profiles: d\bigl(B_0 B_1 B^{(1)},\,B_0 B_1 B^{(2)}\bigr)^{2} \;\propto\; \int\bigl|\phi_{B^{(1)}}-\phi_{B^{(2)}}\bigr|^2\,d^3 r. Per-base distances are the building blocks of the whole 64×64 matrix.
Cross-family substitutions (e.g., serine UCC vs serine AGC) differ at two positions, so two per-base distances stack. Because purine-vs-pyrimidine differences are the largest per-base distances in the alphabet (one vortex vs two on a fused ring), any pair with two purine/pyrimidine swaps sits near the top of the distance distribution.
Worked example: leucine CUU vs CUC
Both CUU and CUC code for leucine; they differ only at the third (wobble) position. By simplification 1,
d(\text{CUU}, \text{CUC})^2 \;\propto\; \int\bigl|\phi_U(\vec{r}) - \phi_C(\vec{r})\bigr|^2\,d^3 r.
The U-vs-C base distance has three contributions:
- Ring core: both pyrimidines; both 6-electron rings; both have nitrogens at positions 1 and 3. The toroidal vortex over the ring is similar in shape; magnitudes differ by the NICS ratio between C and U, which DFT places at \mathcal{O}(1) rather than \mathcal{O}(\text{a few}).
- Exocyclic substituent at C4: amino group (–NH₂) on C, carbonyl (=O) on U. This shifts the local dipole magnitude and orientation, and changes the electron density at the spatial location where the next-base stack sees it.
- Position 5: both C and U carry an H at C5 (no methyl — that’s thymine). No additional contribution.
The qualitative prediction: the two leucine codons sit close in stamp space (same first two bases, same pyrimidine class at the wobble), and should be among the most freely interchangeable synonymous pairs across orthologs.
Cross-family example: the six serines
Serine has six codons: UCU, UCC, UCA, UCG (the UCx family) and AGU, AGC (the AGy family). Intra-family distances are all single-position wobble substitutions (small). Inter-family distances involve substitutions at both positions 1 and 2 — U→A and C→G, each a one-vortex → two-vortex transition — so they sit at the high end of the stamp-distance distribution.
The framework’s prediction is sharp: UCx and AGy serines are biologically interchangeable at the amino-acid level but substrate-distinguishable. They should not be evolutionarily interchanged freely in conserved structural contexts; codon usage between the two families should track tissue-specific or structure-specific demands rather than be uniform. Mammalian and bacterial codon-usage tables already show strong context-specific biases between the UCx and AGy serines; the question the metric answers is whether the direction and magnitude of those biases track the predicted stamp distance once tRNA abundance is controlled for.
Cognate anticodon recognition: 64 of 64
The per-base profiles \phi_B also define the cross-bridge pair-interaction energy the base-pairing section builds on. A codon-anticodon mini-duplex at the ribosomal A-site is three stacked Watson-Crick pairs in A-RNA geometry, and its binding energy — in the nearest-neighbor convention biophysics uses for duplex stability — is the sum of three per-pair WC energies, each computable from the same Gaussian-overlap machinery the pair test uses. The framework’s “opposites attract” claim (channel-with-memory) makes a concrete prediction: for every codon, the position-wise complementary anticodon should be the strongest binder among all 64 possible triplets.
The script compute_codon_anticodon.py builds the 64×64 codon-anticodon binding matrix from exactly the per-base profiles that pass the pair-level tests and the codon-stamp matrix sanity checks. No parameter tuning beyond the inherited pair model:
| Test | Result |
|---|---|
| Cognate anticodon ranks #1 by binding | 64 / 64 codons |
| Mean selectivity gap (E of 2nd-best minus E of cognate) | +3.33 (arb. units) |
| Wobble (G:U with +2 Å lateral shift at position 2) remains attractive | 16 / 16 codons ending in C |
| Mean codon-anticodon binding, canonical (G:C at position 2) | −10.77 |
| Mean codon-anticodon binding, wobble (G:U with shift) | −7.84 |
| Mean wobble softening | +2.93 (about 27% weakening) |
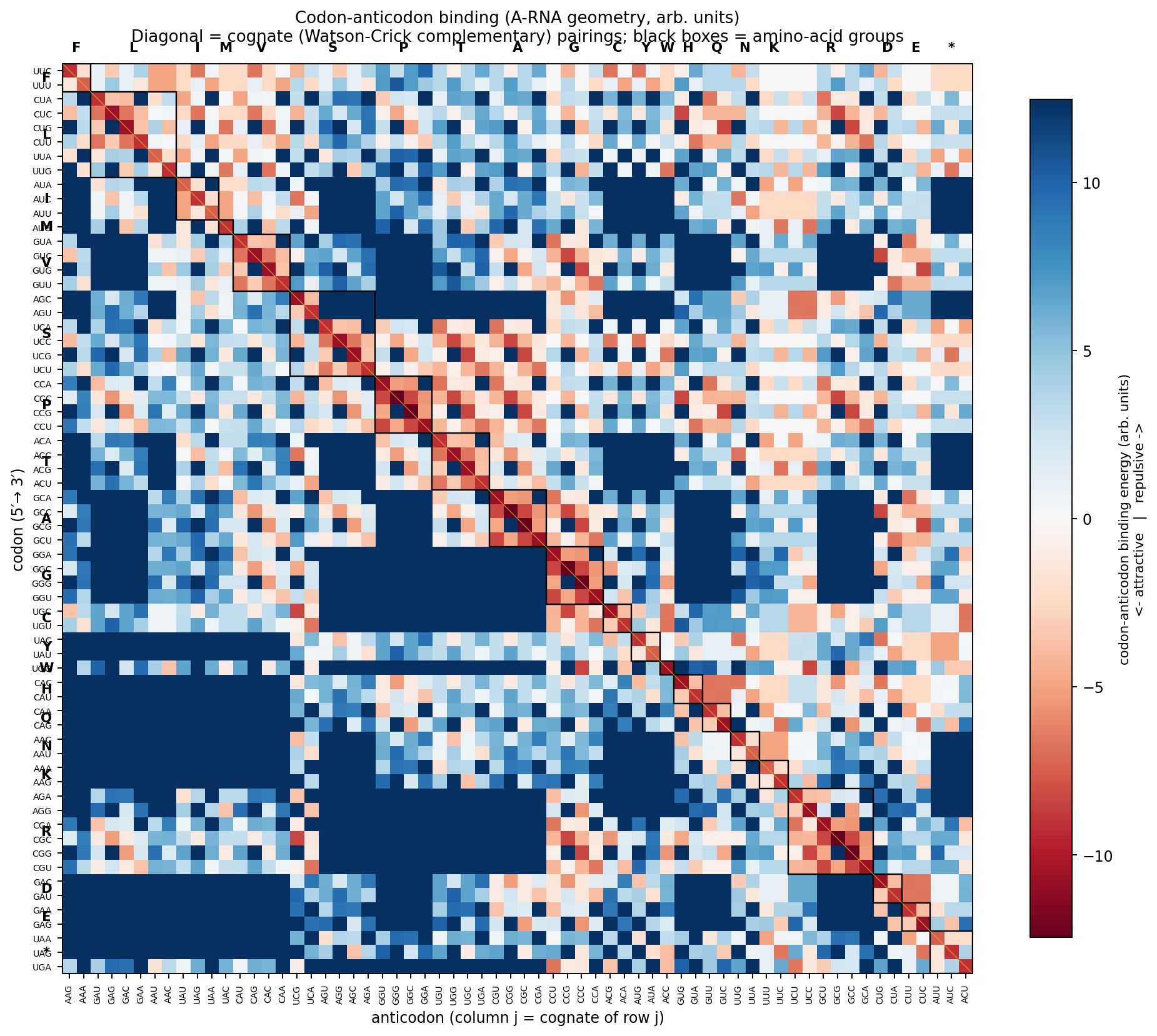
Every codon has a uniquely best partner. The selectivity gap is structural and consistent rather than thermally dominant — a sequence-recognition prediction at the pair level, not an absolute-binding-strength prediction. The wobble extension (G:U pair shifted laterally by ~2 Å, the geometry observed in real wobble pairs) retains most of the canonical G:C binding, dropping from −4.15 (arb.) at G:C to −1.22 at G:U wobble. At the codon-anticodon level the binding weakens from −10.77 to −7.84 — the 27% loss biology tolerates, and the loss the genetic code routes onto the position-3 degeneracy of synonymous codons.
A subtler consequence of binding being a sum of three independent pair energies is that the model is position-blind: a substitution at the 5′ end of the codon and the 3′ end give identical mean energy penalties when averaged across all sequences. The substrate gives the same binding strength to any pair at any stack position. The genetic code’s position-3 degeneracy is therefore not, in this framework, a property of substrate softness at the third base — it is a property of the wobble pair’s own geometric freedom (G:U is allowed to slide laterally; G:C, A:T, A:U are not), and of the genetic code’s choice to route synonymous-codon redundancy onto the position where that freedom is realized. A direct corollary: engineered tRNAs with G:U at the 1st or 2nd codon position should suffer the same binding-energy penalty as at the 3rd position; they are disfavored in biology not because the substrate forbids them but because mistranslating at the first two positions usually changes the amino-acid identity, while at the third position it usually does not.
This sharpens the claim the base-pairing section made on its own. There, the framework noted that “the position in the codon where the substrate’s grip is weakest — the third position, where wobble is allowed — is exactly the position where the genetic code is most degenerate.” The codon-anticodon calculation refines that: the substrate’s grip strength is in fact position-independent at the pair level; what makes position 3 special is the geometric admissibility of G:U, not anything intrinsic about position 3 itself.
Three observables, one set of inputs
The codon stamp and the codon-anticodon binding share a chain of observables, all from one set of per-base inputs with one overall scaling factor:
| Observable | Validation | Source |
|---|---|---|
| Pair-level WC binding, 4 framework propositions | 4 / 4 passing in arb units | validate_pairs.py |
| 64×64 codon distance matrix, 4 sanity checks | 4 / 4 passing; syn / non-syn ratio = 1.75 | compute_codon_matrix.py |
| 64×64 codon-anticodon binding, 3 predictions | 3 / 3 passing | compute_codon_anticodon.py |
Three independent classes of substrate observable — pair energies, codon-stamp similarities, codon-anticodon binding — all derived from one set of per-base profiles with no per-observable tuning. The G:C / A:T ratio (1.69, in the measured 1.8–2.0 range, above the additive H-bond-counting prediction of 1.5), the synonymous codon-stamp distance ratio (1.75), and the codon-anticodon cognate selectivity (64 / 64 rank #1) all emerge from the same inputs.
The pair-level wobble and G:C non-additivity results feed back into the base-pairing section’s framework predictions: the G:U wobble pair recovers from a repulsive +3.50 at canonical placement to an attractive −1.22 with the +2 Å lateral shift — softer than G:C at −4.15, in the predicted direction with no parameter tuning. What remains is absolute calibration (one overall scaling factor against measured pair free energies) before the arb-unit numbers can be compared to experiment in kcal/mol.
The code at the anti-lock pole
A codon does two opposite things at once, and the substrate ladder’s two poles name them. Reading one codon is a locking act: the three stacked vortices nest into a single column — a whole-number stack sharing merged boundary sheets — and the codon-anticodon mini-duplex must bind its cognate — the 64-of-64 result above is a clean lock, a resonance that has to ring. But the 64 codons as a set have the opposite job. The ribosome must never confuse one codon for another, and a system whose failure mode is unwanted confusion between symbols is an anti-lock structure in exactly the sense the retinal cone mosaic is: the eye spreads its cones into a never-periodic packing so incoming spatial frequencies cannot alias into coherent ghosts; a code must spread its codons so a one-letter slip cannot alias one meaning into another. The ladder’s sign rule fixes the pole before any number is computed — name the job, avoid confusion, and the code belongs in the gap, not on a tooth.
Because the 64×64 stamp matrix is already built, this can be tested rather than asserted (scripts/codon-stamp/codon_antilock.py), and the result is a clean mixed verdict that sharpens the claim more than a flat confirmation would.
The lock side holds. Synonymous codons sit close — mean stamp distance 0.18 against 0.32 for codons of different amino acids (ratio 1.74; a shuffle of the amino-acid labels never reaches that proximity, p < 10^{-3}). Where confusion is safe — two spellings of one amino acid — the code lets the stamps lock together.
The substrate hands biology an alphabet it cannot fully separate. A within-class single-base swap — a transition, A↔︎G (purine↔︎purine, two vortices each) or C↔︎U (pyrimidine↔︎pyrimidine, one each) — costs a stamp distance of only \sim\!0.05, while a cross-class transversion (one vortex against two) costs \sim\!0.18, a factor of 3.3 larger. The purine/pyrimidine “one vortex versus two” gap is the alphabet’s metric, and it means roughly half the single-letter swaps the substrate can make are nearly invisible to it.
The code routes its confusions onto exactly those near-invisible swaps — the anti-lock pole’s only available move when the alphabet itself is too cramped to separate everything. Synonymous single-letter neighbours are 46\% transitions against a 33\% baseline, and among the thirty closest codon pairs in the whole matrix, synonymous pairs are over-represented sevenfold (37\% versus 5\% overall). The substrate’s worst indistinguishabilities are spent where they do no harm. This also dissolves the apparent competition in prediction 3 below: the code’s well-known transition robustness and its stamp similarity are not rival explanations — transitions are the small stamp distances, so a transition-robust code and a stamp-routed code are the same code seen twice.
Not every silent mutation is equally silent — and that might be testable. Split the synonymous swaps themselves by type and the two halves separate as cleanly as the alphabet did: a silent transition costs a stamp distance of 0.055, a silent transversion 0.199 — a 3.6\times gap, with nearly every case sitting at the wobble position (scripts/codon-stamp/codon_antilock.py, T5). The conventional account of synonymous-codon effects runs through translation speed — rare codons slow the ribosome and reshape co-translational folding — and that account is blind to this split, because tRNA abundance is a property of which codon while transition-versus-transversion is a property of the change: two silent swaps from one codon to two equally-frequent alternatives can be one of each, the same speed effect carrying a 3.6\times different stamp. So the framework predicts a residual the speed model does not — among silent variants, transversions should carry the larger functional consequence per event, orthogonal to codon usage — and because it is a ratio of stamp distances, it survives the one unset scaling factor untouched. Prediction 2 below made this the chapter’s cleanest already-runnable test; it has now been run against ClinVar — and the honest verdict is negative. The apparent enrichment dissolved confound by confound (allele frequency, then splicing, then a 57%-protein-altering transcript-discordance contamination, then CpG mutability); in the one clean comparison — genuinely synonymous on MANE, splice-neutral, frequency-matched, non-CpG — transversions show no excess (\text{OR}\,0.97 [0.67–1.41]). The signal’s residence only at CpG, a property orthogonal to the stamp, marks it mutational rather than structural.
But the code is not the cone mosaic, and the test says so plainly — which is where it stops being a re-description. A blue-noise packing spreads every element more evenly than random; the codon cloud does the opposite, with nearest-neighbour distances less even than a matched random cloud (coefficient of variation 0.51 against 0.27), because the synonymous families clump. The reason is structural and worth stating: the retina has no synonyms — every cone must be distinguishable from every other, so a uniform spread is its only solution. The code has degeneracy. It does not need all 64 codons mutually distant, only the meaning-changing pairs distant, so it never solves the packing problem the retina solves. It converts a packing problem it cannot win — too few letters, transitions too cheap — into a labelling problem it can, clustering the safe confusions and leaving the dangerous ones as spread as the cramped alphabet allows. Same pole, same sign rule; a different tool, because the code has one the eye lacks.
The residual is where that spread fails, and the test names it. The closest meaning-changing pairs are all transitions the substrate cannot separate and synonymy could not absorb: Thr↔︎Ile (ACA/AUA), Thr↔︎Met (ACG/AUG — and AUG is the start codon), Ala↔︎Val (GCA/GUA), Lys↔︎Arg, Lys↔︎Glu. These are the transition-biased, mistranslation-prone pairs the ribosome-fidelity literature already tracks; the framework reads them not as accidents but as the unavoidable residue of an alphabet whose within-class swaps the substrate renders near-identical. The genetic code is thus the sign rule reaching into the paper’s own biology — not a fourth chemistry-free witness of the gap (it is chemistry), but a demonstration that the avoid-confusion job lands at the anti-lock pole even when the implementer is a molecular code rather than a meristem, a retina, or a buried clock.
What this still predicts (and what would falsify it)
The three observables above are computed; their qualitative directions and orderings land where the framework expects. Three further predictions extend the picture into evolutionary and biomedical data:
Synonymous codon usage bias correlates with stamp distance. Codons separated by larger stamp distances should show stronger context-specific usage; codons close in stamp space should be more freely interchanged. The correlation should survive after controlling for tRNA-abundance effects.
Synonymous SNPs disrupting protein function track stamp distance — and silent transversions bite hardest. “Silent” mutations cause disease at a rate that is not zero. The framework predicts that the disease rate per synonymous substitution should correlate with the stamp distance between original and substituted codon, not just with codon-usage frequency — and the transition/transversion split sharpens this into a test that can be run on public data today: silent transversions (d \approx 0.20) should be enriched roughly 3.6\times over silent transitions (d \approx 0.055) among functionally-consequential silent variants. The ClinVar dataset did not show this pattern for various reasons but MaveDB protein-abundance MAVEs with synonymous coverage might show this pattern.
The genetic code’s non-randomness reflects stamp similarity because stamp similarity is what error-minimization minimizes. The codon table groups physicochemically similar amino acids near each other in codon space. The standard reading calls this error-minimization (the code is robust to mutation, especially to transitions); the framework’s anti-lock analysis shows these are not competing explanations but one — transitions are the small stamp distances (within-Hückel-class swaps, \sim\!3.3\times closer than transversions), so a transition-robust code is a stamp-routed code. The sharpened, falsifiable prediction: across orthologs and across the standard amino-acid property scales, residual codon-correlated variance should track stamp distance — and in particular transversion substitutions should carry more functional consequence per event than transitions beyond what hydropathy/volume scales alone predict, because the substrate makes transversions the distinguishable swaps.
The picture is falsified if, with chemistry-only inputs and one scaling factor, the 64×64 distance matrix shows no correlation with synonymous-codon usage variation across orthologs once tRNA abundance is controlled. It is supported if the correlation appears and matches the predicted direction. The calculation is concrete; the data is public; the answer is binary.
Honest assessment
What is solid at this stage: the framework, the per-base inputs (all five bases have published NICS, electron density, and dipole data), the convolution geometry, the metric definition, and three independent classes of substrate observable — pair-level WC binding, the 64×64 codon-stamp distance matrix, and the 64×64 codon-anticodon binding matrix — each landing in the framework-predicted direction without per-observable tuning. The substrate prediction holds at the level of qualitative orderings and directional energy gaps; absolute magnitudes still ride on one unset scaling factor.
What is not yet solid:
- The absolute magnitudes of the distances. One scaling factor must be set from data; nothing in the framework currently pins it.
- The precise pairwise ordering when per-base differences are small (within-pyrimidine and within-purine distances).
- Whether the small magnitude of synonymous-codon stamp differences is enough to bias evolution against the much larger noise from tRNA abundance, mRNA secondary structure, and translation context. This is the substantive open question — and the direct confrontation (silent transversions in ClinVar) came back negative once all four confounds were controlled. The apparent enrichment was peeled away in stages — allele frequency, splicing (excluded by SpliceAI), a 57%-protein-altering transcript-discordance contamination, and finally CpG mutability — leaving no excess in the clean comparison (genuinely synonymous on MANE, splice-neutral, frequency-matched, non-CpG: \text{OR}\,0.97 [0.67–1.41]). ClinVar cannot give this prediction a clean hearing; only a splice-free direct assay (deep-mutational-scanning of protein abundance, with synonymous coverage) could, carrying none of the four confounds. The matrix’s computed observables are unaffected — this closes one downstream prediction, not the stamp construction.
- The geometry choice (B-form storage vs A-form ribosomal A-site). The qualitative metric should be robust across both, but a quantitative result requires committing to a geometry; both passes are worth running.
The codon stamp is offered here as a falsifiable proposal whose three computed observables already land in the predicted direction. The remaining follow-up is mechanical: run the 64×64 matrix against published codon-usage data across orthologs, and against the AlphaFold-style structural datasets that look for residual codon-correlated variance at each amino-acid position. The pre-substrate ribosomal-RNA and tRNA-folding observations — patterns suggesting structural codon-stamp matching on rRNA stems and tRNA L-shapes — would, if the stamp works, finally have the physical model they were always missing.